

Peras con manzanas
Si tuviésemos unos lentes que permitieran amplificar al menos mil veces lo que vemos, veríamos pequeños puntos en todas las superficies, en el aire, en los líquidos.
Esos puntos son microorganismos. Como no podemos verlos a simple vista, vivimos nuestro día a día como si no existieran... Hasta que en la cita médica nos enteramos de la infección a la faringe y tenemos que suspender el pisco sour y tomar antibióticos.
Pero aunque no los veamos ni nos produzcan una infección, esos organismos microscópicos siempre están ahí y surgieron en este planeta muchísimo antes que cualquier otra criatura con orejas o patas.
Los seres vivos se parecen entre sí. Todo depende del carácter que usemos para esas comparaciones. Podríamos agrupar a todos los organismos que tienen escamas o aquellos que tienen ojos y formar grupos específicos y los clasificamos de acuerdo a esos caracteres. Es de esta clasificación que surge el clásico «árbol de la vida», donde se puede ver cómo están emparentados todos los seres vivos que conocemos en el planeta.
Pero, ¿cómo hacemos para discernir entre una especie y otra? Actualmente, lo que más se usa son sus diferencias genéticas. Es decir, qué tan distinto es el alfabeto que nos escribe a cada uno. Sin ir más lejos, uno de nuestros etilmercurianos encontró un gen del piure (esa cosa que parece una roca ensangrentada y que hay gente que —guácala— se come) que es un 95,5% similar a un gen humano y... Momento. ¿A quién le importan los piures?
Filo filo, filo contigo

Hace unos días, investigadores del Australian Center for Ecogenomics publicaron el trabajo «Recovery of nearly 8,000 metagenome-assembled genomes substantially expands the tree of life» (1). Como dice el título, usando bases de datos públicas y potentes herramientas computacionales, el equipo de investigación consiguió «ensamblar» casi 8 mil genomas [conjunto de genes de todo el organismos, los cuales a su vez están formados de ADN] de microorganismos (Archaea y Bacteria). De ellos, 20 corresponden a organismos que no estaban descritos anteriormente a nivel de filo (o phylum en latín). En taxonomía, filo es un nivel superior de clasificación de organismos; luego (y si ignoramos categorías intermedias más específicas) vienen clase, familia, género y especie.
Es decir, cada nuevo genoma descubierto por este grupo representa un nuevo filo bacteriano; en este filo pueden existir cientos de «especies» distintas, desconocidas hasta ahora (así que imagínese todo lo que falta por descubrir). Solo este estudio contribuyó a aumentar en un 30% el número de filos existentes en el árbol de la vida.
Pero, ¿de dónde sacó el grupo de investigación los datos para «ensamblar» genomas? ¿Y cómo se puede recuperar la información genética de organismos tan pequeños?
Bring me the data
Como los microorganismos son criaturas unicelulares, difíciles de analizar, el uso de caracteres taxonómicos (escamas, ojos, esqueleto... o sea, las características «observables» que nos permiten diferenciar un organismo de otro) no es suficiente para discriminar entre grupos. Por esto, se hace necesario obtener la información genética de estos organismos junto con otro tipo de información (bioquímica, fisiología).
El otro problema que tenemos es que, como dijimos anteriormente, la mayoría de los microorganismos no son cultivables. Eso significa que la única forma de acceder a ellos es «recoger» los genes que están en el ambiente. Sin embargo, los microorganismos que pueden ser cultivados siguen siendo útiles, ya que se les ha secuenciado su genoma (todos sus genes) y así se convierten en los «genomas de referencia» para aquellos organismos no cultivables (si quiere saber más sobre esto, puede visitar nuestro post sobre cuánto pesa su genoma).
¿Qué es una especie? ¿De dónde venimos? ¿Para dónde vamos? ¿Dónde está el quinto dedo en los perros? ¿Por qué le estoy revisando las patas a mi perro ahora? Imagen: Sergio Melnick

Definir qué es una especie en organismos «superiores» es complejo y aún es objeto de debate. De hecho, hay más de 40 definiciones (acá, un blog amigo recopila al menos 26 definiciones distintas). Ahora, pensando en microorganismos donde es complejo establecer diferencias, esto se torna aún peor. Incluso la existencia del concepto de «especie» es difícil de delimitar. Por este motivo se habla de «filotipo» o «unidad taxonómica operacional» (OTU, por su sigla en inglés), lo cual está basado en la similitud de la secuencia del gen en estudio con la información que existe de otros organismos.
La información genética (secuencias) que se obtiene mediante distintas técnicas como clonamiento y secuenciación directa masiva es almacenada en bases de datos públicas y abiertas como NCBI-GenBank, EMBL-EBI y otras como Sequence Read Archive (SRA) para datos masivos. Entre los años 1990 y la primera década del 2000, la técnica más usada era la clonación, mediante la cual se obtiene por «corrida» miles de secuencias de ADN, lo que equivale a cerca de 700 Mb de datos. Es una cifra no menor si tomamos en cuenta que esos ~700 Mb de datos son «solamente» 4 letras (¡no sea ordinario!) empalmadas una detrás de la otra en distinto orden. No hay otro tipo de información contenida en estas secuencias de ADN.
Actualmente, las técnicas de secuenciación masiva producen entre 10-1000 Gb por corrida. Por ello, gran parte del trabajo que se hace en biología y genética consiste básicamente en bucear en estas enormes bases de datos.
Dentro del estudio de las comunidades microbianas en el ambiente, son frecuentes los trabajos sobre diversidad microbiana usando uno o algunos genes marcadores. Es decir, genes que sean informativos. Dependiendo de la pregunta es que definimos qué significa informativo. Para este tipo de estudios, generalmente se utilizan aquellos que son conservados a través del tiempo, o que tienen una baja tasa de mutación al azar. Esto nos permite evaluar aquellas diferencias a las que llamamos «profundas», que nos ayudarán a trazar límites (genéticos) entre un grupo y otro. Para esto, el gen más común es uno que se encuentra dentro de los ribosomas llamado 16S. O simplemente el gen ribosomal 16S.
¿Cuántos genes hay en mi patio?

Gracias al avance tecnológico de secuenciación masiva de última generación (NGS para los amigos), cada vez es más frecuente analizar todos los genes presentes en una muestra ambiental o, como le llamamos en ciencia, todo el ADN metagenómico. Ya no nos referimos solo a un gen como lo hacíamos en el pasado. Esto se puede hacer de forma muy simple: si nos interesa, por ejemplo, estudiar los genes que hay en el suelo del patio de la casa (si vive en departamento, imagine el macetero de su cactus), se toma una muestra y, literalmente, extrae su ADN. Existen kits comerciales que nos ayudan en este trabajo: se trata de un set de reactivos que rompen la pared de las células y liberan el ADN al medio: este ADN puede ser purificado y analizado. El ADN puede secuenciado directamente en distintas plataformas de secuenciación generando decenas de Gb de información. Es decir, el ADN es leído y organizado de acuerdo a sus genes. El ADN obtenido debe analizarse cuidadosamente usando herramientas y software especializados (la mayoría son de acceso y código libre, compañero).
Lo primero que se hace es clasificar las secuencias, es decir, saber a qué se parecen. Dependiendo de su similitud, se le asocia un gen que ya haya sido descrito (esto no siempre es posible: todo depende de la cantidad de información que otros grupos de investigación hayan generado). Si logramos esto, podríamos saber que en el suelo del patio hay genes relacionados a la fijación de dióxido de carbono, por ejemplo, o a la resistencia a antibióticos. Para que esto ocurra es necesario acceder a bases de datos actualizadas sobre clasificación taxonómica de microorganismos, y existen muchas de ellas. Entre las más usadas actualmente están SILVA, Greengenes (no, no tiene que ver con Greenpeace), Ribosomal Database Project (RDP), entre otras.

¡TODO NIÑO TIENE DERECHO A QUE LA INFORMACIÓN SEA DE ACCESO LIBRE!
Acceder a estas bases de datos es crucial para el avance de la investigación del Microbioma, es decir, el estudio de los microorganismos que viven en un determinado ambiente. Debido a la gran magnitud de diversidad microbiana en el planeta, la única forma de conocerlos es teniendo una base de datos robusta e información de buena calidad que permita hacer comparaciones. Por lo tanto, no tiene sentido que exista información «privada» o «confidencial» en este tema. Al contrario: el acceso libre a la información es fundamental para el avance de la ciencia.
Pasa muchas veces que no existen genes conocidos para algunas secuencias y éstas se denominan «unassigned» (no asignadas). En el caso de que la secuencia se asocie a un gen, pero no se puede determinar a qué organismo se parece, se le llama «unclassified» (no clasificada).
Tanta información genética equivale a tratar de armar un rompecabezas de millones de piezas. Manualmente, sería casi imposible encontrar algún patrón a seguir: por esto es fundamental el uso de herramientas computacionales robustas.
Nuevamente, basado en algoritmos de similitud de secuencia y complementariedad, todos los genes encontrados se «ensamblan», tratando de reconstituir un genoma ya conocido. El genoma refleja todos los genes de un organismo, por lo que si tenemos acceso al genoma, podemos predecir o inferir qué organismo está (o estuvo) presente en un determinado lugar. El ADN es una molécula muy estable, que deja huellas (casi) imborrables. Imborrables como lo que no se puede borrar.
Entonces, si se secuencia el ADN proveniente de un poco de tierra, unos cuantos mililitros de agua o incluso el aire (concentrando el viento con filtros), este puede ser ensamblado. Dependiendo de qué tan profunda es la secuenciación, será posible poder ensamblar genomas desde datos metagenómicos. Esto complejiza el rompecabezas; pero, afortunadamente, hay muchas piezas similares que encajan bien entre sí y existen herramientas que nos ayudan a predecir cómo ensamblarlas.
En décadas pasadas, el problema era generar los datos. Ahora es justo al revés: se obtienen tantos datos que procesarlos requiere una capacidad humana que supera a proyectos específicos y a muchas instituciones de investigación científica. Lo ideal sería entonces generar iniciativas gubernamentales de grandes repositorios y bases de datos, como ya se ha hecho en Estados Unidos y la Unión Europea. E incluso con esos recursos, realizar el análisis de los datos es un desafío titánico. Por eso, el logro del Australian Centre for Ecogenomics no es algo menor.
Muchas veces se habla de Big Data refiriéndose principalmente a datos astronómicos o de información generada por redes sociales. Pero los datos genómicos también son Big Data... Que crece a una escala exorbitante y que requiere de adquisición, almacenaje, distribución y análisis (2).
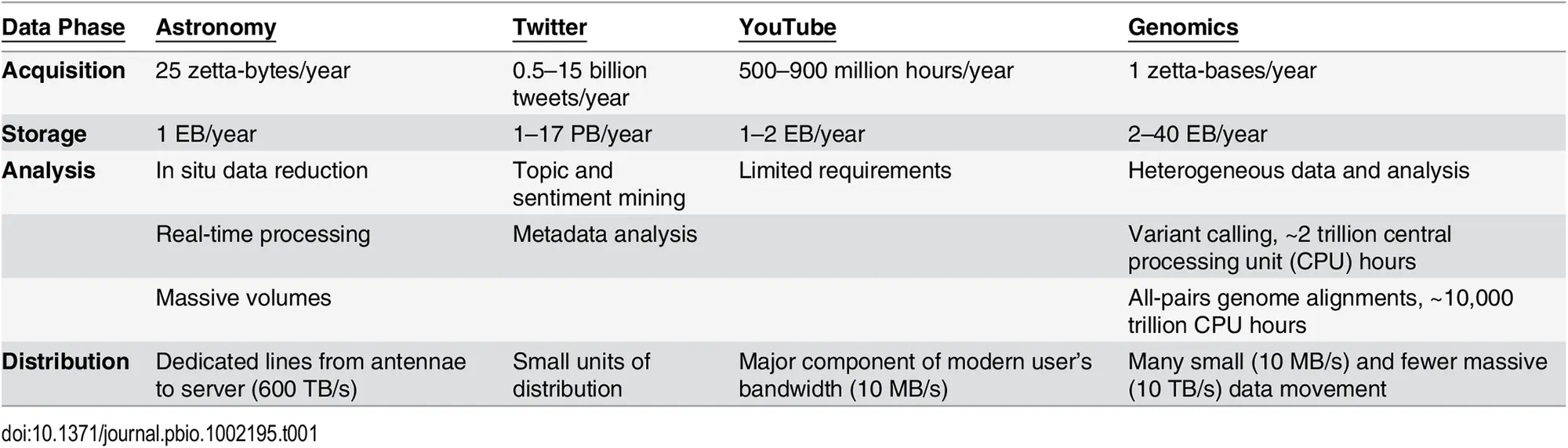
Cuatro dominios de Big Data para el año 2025 (2)
El árbol de la vida se convierte en un bosque
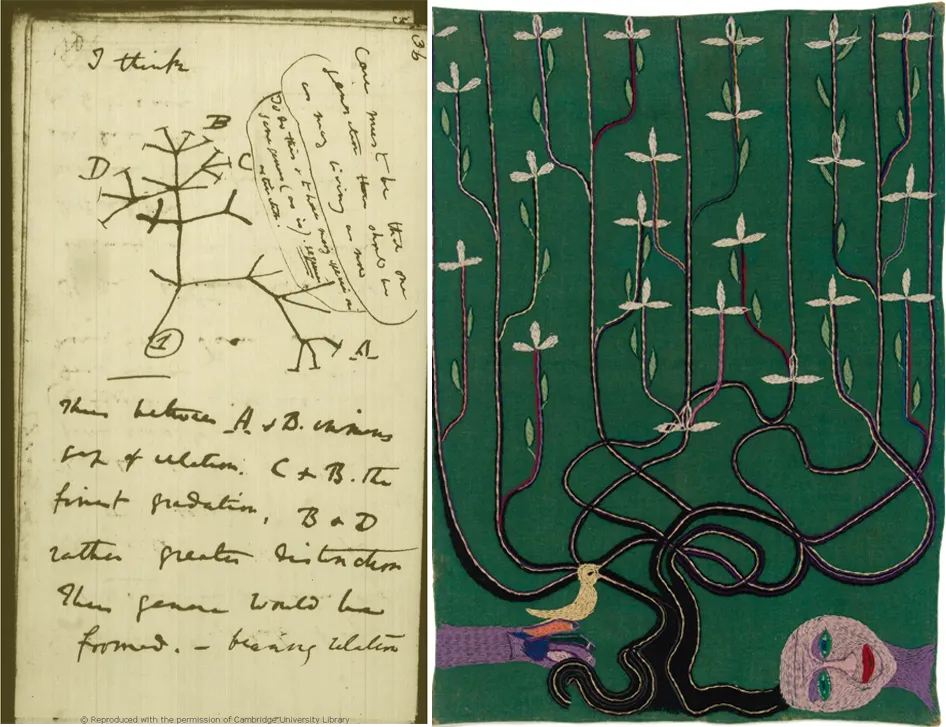
El árbol de la vida según Darwin (1837) y Violeta Parra (1963) <3
El equipo del Australian Centre for Ecogenomics no se dedicó a recoger muestras de las macetas de sus cactus, sino que accedieron a las bases de datos públicas para ensamblar sus casi 8 mil genomas de Archaea y Bacteria, descubriendo nuevos grupos en el árbol de la vida. O sea, no fue necesario hacer un estudio multimillonario a nivel global, sino que se «exprimieron» datos ya existentes, a los que han contribuido investigadores de todo el mundo. El trabajo de los australianos, sin embargo, es destacable: tuvieron que tener acceso a una enorme capacidad computacional para analizar miles de millones de secuencias. Es por esto que el gran desafío de la biología en el futuro se encuentra en los datos genómicos. Si le gusta la computación y la bioinformática, las herramientas y los datos para hacer ciencia están a su disposición.
Los resultados de este trabajo complejizan aún más nuestra visión de la diversidad biológica microbiana. Ya no es discusión para nadie que los microorganismos son los seres vivos más diversos y abundantes del planeta, pero aún queda un universo microbiano por descubrir. Lo cierto es que este árbol de la vida de tres dominios, con unas pocas ramas, presentado en 1990 por Carl Woese y sus colegas (3), se ha convertido en un cada vez más complejo bosque que crece a un ritmo acelerado.
Ahora, y yendo un poco más lejos, hay investigadores que han descubierto que la distancia filogenética entre Archaea y Eukarya es cada vez menor, principalmente porque los eucariontes se originaron por la fusión entre células de Archaea y Bacteria (4; 5; 6). Por ello, se ha propuesto la existencia de solo dos dominios de la vida: Archaea y Bacteria, siendo Eukarya un grupo hermano de Archaea. Esta idea ha causado revuelo ya que a nivel celular serían grupos distintos (7).
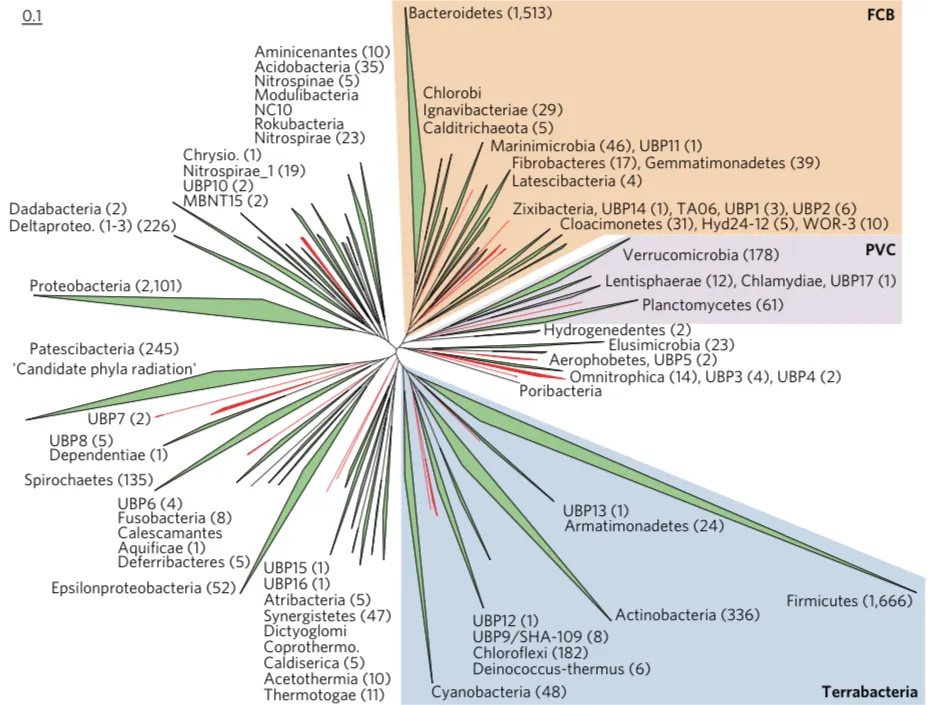
Presencia de los nuevos linajes descubiertos de Bacteria en el árbol de la vida (1)
Pero, independiente del consenso que surja de esa discusión, lo fascinante es que los humanos seguimos ocupando una ramita ínfima en el frondoso bosque de la vida.
Referencias
1.
Parks DH, Rinke C, Chuvochina M, Chaumeil P-A, Woodcroft BJ, Evans PN, Hugenholtz P, Tyson G (2017) Recovery of nearly 8,000 metagenome-assembled genomes substantially expands the tree of life. Nature Microbiology doi:10.1038/s41564-017-0012-7
2.
Stephens ZD, Lee SY, Faghri F, Campbell RH, Zhai C, Efron MJ, et al. (2015) Big Data: Astronomical or Genomical? PLoS Biol 13(7): e1002195. https://doi.org/10.1371/journal.pbio.1002195
3.
Woese CR, Kandler O, Wheelis ML. Towards a natural system of organisms: proposal for the domains Archaea, Bacteria and Eucarya (1990). Proc Natl Acad Sci USA 87:4576-4579
4.
Williams TA, Foster PG, Cox CJ, Embley TM. An archaeal origin of eukaryotes supports only two primary domains of life (2013). Nature 504:231–236
5.
Koonin EV. Origin of eukaryotes from within archaea, archaeal eukaryome and bursts of gene gain: eukaryogenesis just made easier? (2015) Philos Trans R Soc Lond Ser B Biol Sci. 370:20140333
6.
Raymann K, Brochier-Armanet C, Gribaldo S. The two-domain tree of life is linked to a new root for the archaea (2015). Proc Natl Acad Sci USA 112:6670–5
7.
van der Gulik PTS, Hoff WD, Speijer D (2017) In defence of the three-domains of life paradigm. BMC Evolutionary Biology 17:218